
Welcome to the essential yet often behind-the-scenes world of regulatory medical writing. This field stands at the crossroads of science and communication for the approval of new medical treatments.
Regulatory writers are the linchpins in this process, tasked with translating complex clinical data into clear, compliant documents for government agencies.
Let’s look into the intricacies of what regulatory writers do and their critical role in advancing medical science.
What Is Regulatory Medical Writing?
Regulatory medical writing is a specialized domain within the broader field of medical writing, focusing on creating documentation required by regulatory agencies to approve drugs, biologics, and medical devices.
Imagine medical writing as a bridge facilitating dialogue between pharmaceutical companies, biotech firms, medical device manufacturers, and regulatory agencies such as the FDA (Food and Drug Administration) in the United States, EMA (European Medicines Agency) in Europe, and other regulatory bodies worldwide.
Regulatory medical writers play an important role in the drug development process and regulatory affairs, working closely with scientists, researchers, and healthcare professionals to guarantee that all clinical documents and submissions meet the regulatory guidelines and standards set by these agencies. This involves the preparation of a wide array of documents, including, but not limited to, clinical trial protocols, clinical study reports, patient-informed consent forms, summary documents, and marketing application dossiers.
But here’s the kicker — the regulatory environment is highly dynamic, with regulations and guidelines continuously evolving to adapt to new scientific discoveries, technological advancements, and changes in public health policy. So, regulatory medical writers have to stay on top of these changes to ensure compliance and facilitate the successful navigation of the regulatory process.
Working within a medical writing team, a regulatory writer contributes significantly to the strategic planning and execution of the regulatory submission strategy.
Their expertise makes sure the drug development process progresses smoothly and that the documentation accurately reflects the research findings, supporting the safety and efficacy of new drugs or medical devices. And that’s something to talk about!
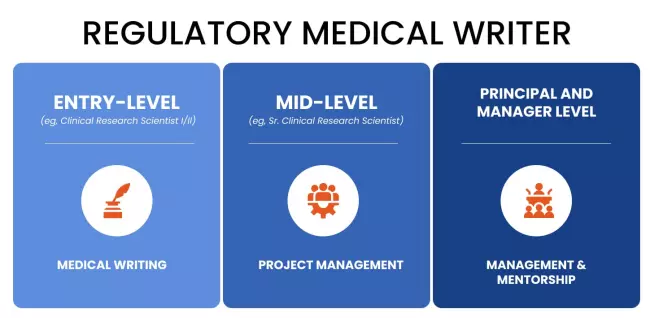
What Does a Regulatory Medical Writer Do?
There are different types of regulatory medical writers, each having different tasks.
Entry-level (eg, Clinical Research Scientist I/II): Medical Writing
As an entry-level regulatory medical writer, the most time is spent analyzing and summarizing data from clinical trials for various audiences, but predominantly regulatory authorities (eg, the FDA).
Entry-level writers often start out writing Clinical Study Reports (CSRs) that summarize the design, conduct, and results of a clinical trial. (For those interested in seeing a CSR’s structure, click here.)
Typical activities of an entry-level writer may include:
- Summarizing safety and/or efficacy data for a CSR
- Incorporating comments from your Project Manager or Project Team
- Proposing a project timeline at a team meeting
- Participating in a roundtable meeting (sometimes called comment resolution meeting)
- Getting exposure to other regulatory documents (via training or working with a more senior writer)
- Performing a quality control (QC) check of a fellow writer’s CSR
What is QC, you ask? Quality control of a document involves a 100% data check, as well as checking grammar, consistency of language, and adherence to a style guide (guidelines to define the style of writing for a particular compound, ensuring consistency across documents). Some companies, like Syner-G, have their own QC department, where in other companies, QC is performed by medical writing peers. Attention to detail is crucial here.
Mid-Level (eg, Sr. Clinical Research Scientist): Project Management
As a writer’s career develops, the focus shifts to project management as well as writing more complex regulatory documents (eg, components of a New Drug Application [NDA]).
Typical activities of a mid-level writer may include:
- Reviewing documents written by entry-level writers and providing constructive feedback
- Attending conference calls to discuss messaging across documents within an NDA
- Participating in a team meeting to shorten timelines for a critical document
- Discussing budget, timeline, or resource needs with a client
- Leading a roundtable meeting to discuss a client’s review of a document
Client meetings require a mix of diplomacy and regulatory experience to achieve one goal: provide the highest quality document to the client in (usually) the shortest time possible.
The ability to be efficient, organized, and flexible are translatable skills from many different backgrounds that will serve you well as a regulatory medical writer.
Principal and Manager Level: Management and Mentorship
In addition to writing and project management, as your career progresses, other opportunities become available. Depending on your interests, this could include line management and/or managing complex projects or programs.
Line managers oversee the resourcing of projects and provide mentoring/career development guidance so writers gain exposure to new and different types of projects.
At this level, medical writers may act as project managers over an entire NDA (or the European equivalent, a Marketing Authorisation Application [MAA]) or manage other large or complex regulatory writing projects.
Where your career goes from here depends on you and your individual abilities and preferences. Cool stuff, huh?
The Importance of Regulatory Medical Writing in the Approval Process
Regulatory medical writing plays a pivotal role in the healthcare sector, particularly in the drug and device approval process. Its significance cannot be overstated, as it bridges the gap between innovation and patient access. Here are key points highlighting its importance:
Ensuring Regulatory Compliance
Regulatory medical writers prepare and submit documentation essential for the approval of drugs, devices, and biologics, ensuring that these submissions meet the stringent requirements set by regulatory agencies.
Efficient and compliant regulatory writing accelerates the review process, reducing delays and facilitating the quicker introduction of new therapies.
Facilitating Clear Communication
Regulatory medical writing creates a transparent and effective communication channel to address all regulatory queries accurately, reducing the likelihood of submission rejections or requests for additional information.
It also produces clear and accessible documentation, such as product labels and patient information leaflets, which is crucial for the safe and effective use of new medical products.
Translating Science into Solutions through Regulatory Medical Writing
Regulatory medical writing is the unsung hero in the pharmaceutical world, translating scientific discoveries into approved treatments.
These writers bridge the gap between complex clinical data and regulatory standards, crafting documents essential for government agency approvals. Their meticulousness makes sure new drugs and medical devices can navigate the approval process, contributing to advancements in healthcare and patient well-being.