
In the ever-evolving landscape of cancer treatment, timely access to effective therapies is crucial. The FDA’s Real-Time Oncology Review (RTOR) program is a groundbreaking initiative designed to expedite the review process for oncology drugs and biologics ensuring that patients receive life-saving treatments as quickly as possible.
Overview
A major challenge with the FDA’s traditional review process is the lengthy review times. Factors that contribute to this challenge are encountering unexpected safety and efficacy concerns during review, and issues related to manufacturing (quality control or consistency).
The Oncology Center of Excellence Real-Time Oncology Review (OCE-RTOR) program was created to address these issues. Initiated in 2018, the RTOR program aims to provide a more efficient and timely review process to ensure that safe and effective treatments are available to patients as early as possible. Maintaining and improving review quality and balancing the review team’s workload is accomplished through data and analysis standardization, and early iterative engagement with the applicant.
RTOR facilitates the early submission of topline efficacy and safety results, prior to the submission of a complete New Drug Application (NDA) or Biologics License Application (BLA), to support an earlier start to the FDA’s evaluation of the application.
Scope
The RTOR is particularly focused on treatments that have limited or no available therapies, addressing urgent patient needs. When the pilot program was rolled out, supplemental applications were preferred/considered. Since then, the program has evolved to consider NDAs and BLAs for new molecular entities along with their respective supplemental applications.
Participation in this program is voluntary, and acceptance into RTOR does not guarantee or influence approval of the application. Criteria for Acceptance of NDAs or BLAs are as follows:
- Demonstration of substantial improvements over available therapy,
- Straightforward study designs determined by FDA’s respective review Division and the OCE,
- Easily interpretable end points.
It is to be noted that the Prescription Drug User Fee Act (PDUFA) review clock starts once the application is considered complete, i.e., when the final component is received by the FDA.
Timeline and Process
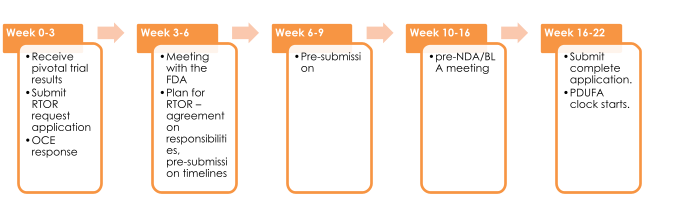
The following figure provides a snapshot of the standard operating procedure for submission made under RTOR program. These milestones are provided as a general guideline and may vary across applications.
Pre-Submission Content
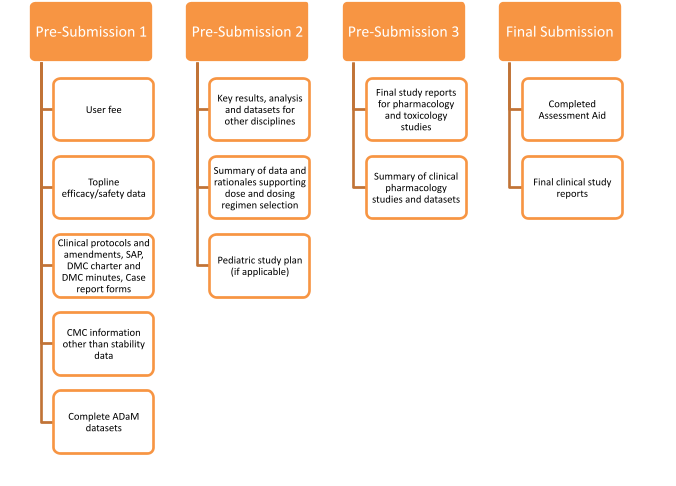
The components of the marketing application can be bundled into a maximum of three pre-submissions and a final submission. An example of RTOR pre-submission content can be found below.
How can you align Quality (CMC) with RTOR
Often chemistry, manufacturing, and controls (CMC) development timelines do not align with the clinical timelines. Since eligibility into RTOR is constrained by availability and submission of clinical data, it becomes important to align CMC development activities with RTOR timelines to make the most out of the program. Some key points applicants can keep in mind while interacting with the FDA on the CMC aspects of the applications are summarized below:
- Encourage more flexible approaches to ensuring information exchange and understanding to facilitate expediting development and review.
- Agree upon a schedule of important review milestones and turnaround timeframes for information requests.
- Discuss approach to submit agreed-upon data packages during the review:
- In the case of small molecules, submission of the dissolution method development report and dissolution specification setting strategy for early review by FDA Biopharmaceutics reviewers
- Additional real-time stability data on the proposed commercial product,
- Additional batch data to support validation.
- Initiate discussions to enable rapid access to CMC and facility data to facilitate pre-approval inspection scheduling and conduct.
Conclusion
- Good Initiative
This initiative is commendable as it aims to improve processes and outcomes. By introducing this program, it shows a proactive approach to addressing current challenges and enhancing efficiency.
- Benefit – Allows Early Access to Data and Opportunity for Review and Consult
One of the primary benefits of this initiative is that it provides early access to crucial data. This early access enables stakeholders to review and consult on the data promptly, leading to more informed decision-making. It can also help identify potential issues early on, allowing for timely interventions and adjustments.
- Burden to the Applicant?
While the initiative has its advantages, it may also impose certain burdens on the applicants. These could include additional documentation requirements, more stringent timelines, and the need for more frequent updates. Applicants might need to allocate more resources to comply with the new requirements, which could be challenging, especially for smaller organizations.
How can Syner-G help?
Syner-G Biopharma Group can assist with RTOR submissions through our comprehensive regulatory strategy and submission services. Here are some ways we can help:
- Regulatory Expertise: We have extensive experience in navigating the complexities of regulatory submissions. We provide strategic guidance tailored to the specific requirements of oncology products.
- Document Preparation and Management: We offer meticulous preparation, review, and management of all necessary documents, ensuring they meet the stringent standards of regulatory bodies.
- Global Submission Support: We ensure that submissions are aligned with local and international standards, facilitating a smooth regulatory review process. This includes authoring and reviewing components for various regulatory agencies.
- Project Management: Our project managers coordinate submission timelines and ensure that all interdependent documents and sections are available in a timely manner, preventing delays.
- Regulatory Agency Interaction: We provide support for meetings with regulatory agencies, helping to prepare strategic questions and comprehensive briefing packages, and participating in these crucial interactions.
By leveraging these services, Syner-G Biopharma Group can help streamline the RTOR submission process, potentially accelerating the approval and market readiness of oncology therapies.
