
The FDA NDA review timeline can be complex, so it’s important to understand it during the drug development process. There are various stages and considerations involved in bringing a new pharmaceutical product to market.
With an emphasis on scientific rigor and regulatory compliance, this article breaks down each phase of the review process, from pre-submission discussions to final FDA actions.
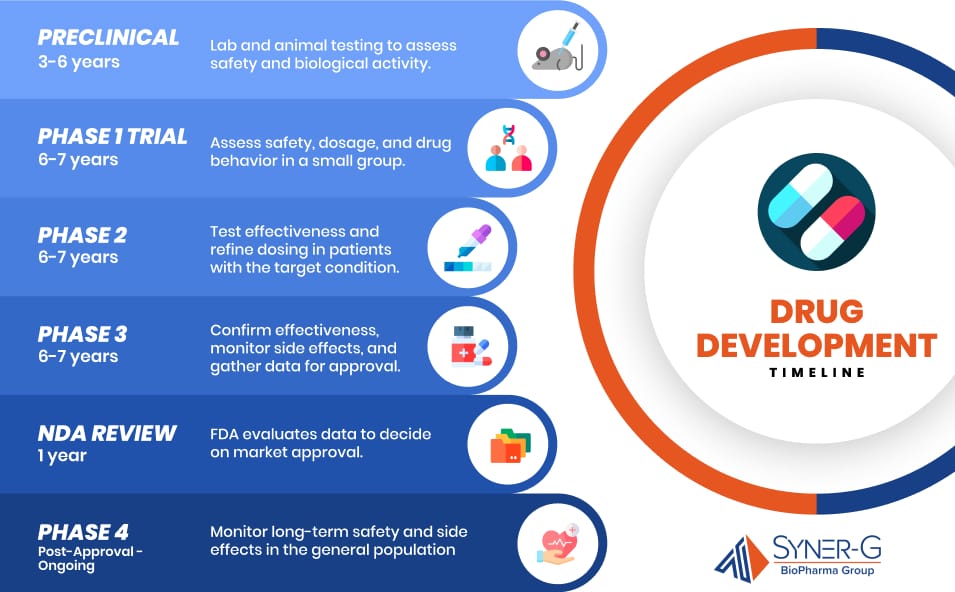
Timeline of Drug Development
Drug development is a complex and lengthy process, encompassing several critical phases that span many years. Each phase is designed to ensure the safety, efficacy, and quality of a new drug before it can be approved for public use. The timeline follows these stages:
- Preclinical (3-6 years): The drug development process begins with preclinical research, where the drug is tested extensively in laboratories and on animals to assess its safety and biological activity. This stage identifies any potential toxicity and lays the groundwork for future clinical trials in humans.
- Phase 1 (6-7 years): Once preclinical testing is completed, the drug enters Phase 1 clinical trials. These trials involve a small group of healthy volunteers or patients and focus primarily on assessing the drug’s safety, dosage range, and pharmacokinetics (how the drug is absorbed, distributed, metabolized, and excreted in the body). This phase, combined with phases two and three, typically last around 6 to 7 years all together, although timelines can vary.
- Phase 2 (Cont. 6-7 years): In Phase 2, the focus shifts to evaluating the drug’s effectiveness in treating a specific condition, as well as further assessing its safety. These trials involve a larger group of patients who have the condition the drug is intended to treat. The goal is to determine the optimal dose and gather preliminary data on the drug’s efficacy.
- Phase 3 (Cont. 6-7 years): Phase 3 clinical trials are the most extensive and involve a larger patient population. These trials are designed to confirm the drug’s effectiveness, monitor side effects, and compare it to standard or equivalent treatments. The data collected during this phase is critical for the eventual New Drug Application (NDA) submission.
- NDA Review (1 year): After successful completion of Phase 3 trials, the sponsor compiles all the clinical and preclinical data, along with manufacturing details, into a New Drug Application (NDA) submitted to the FDA. The NDA review process typically takes about 1 year. During this time, the FDA rigorously evaluates the data to determine whether the drug should be approved for marketing.
- Phase 4 (Post-Approval – Ongoing): Even after a drug is approved and available to the public, it continues to be monitored in Phase 4, often referred to as post-marketing surveillance. This phase is crucial for identifying any long-term side effects or other issues that may arise when the drug is used more widely. Phase 4 can last for many years, ensuring ongoing safety and efficacy in the general population.
Throughout this process, the Investigational New Drug (IND) application plays a central role. Before a drug can enter clinical trials (Phase 1), an IND must be filed with the FDA to obtain approval for human testing.
As the drug progresses through the clinical phases and the data is compiled, the IND eventually transitions into an NDA, which, if approved, allows the drug to be marketed and prescribed to patients.
Submitting Your NDA
Components of the NDA
The New Drug Application (NDA) is a comprehensive dossier submitted to the FDA, encompassing all data necessary to support the approval of a new pharmaceutical product. The NDA comprises several critical sections that provide a holistic view of the drug’s development journey. The clinical section includes data from clinical trials, demonstrating the drug’s efficacy and safety profile.
The non-clinical section provides extensive information on pharmacology and toxicology studies, while the chemistry, manufacturing, and controls (CMC) section outlines the drug’s formulation, production processes, and quality control measures. Together, these components offer a compelling narrative that substantiates the drug’s therapeutic value and its compliance with regulatory standards.
Common Technical Document (CTD) Format
The adoption of the Common Technical Document (CTD) format marks a significant advancement in regulatory submissions, promoting consistency and efficiency. The CTD format standardizes the presentation of data, facilitating a more straightforward and harmonized review process.
This structure is divided into five modules, covering administrative information, overviews and summaries, quality data, non-clinical study reports, and clinical study reports. By adhering to this internationally recognized format, sponsors can streamline their submissions, reduce redundancies, and enhance the clarity and accessibility of their data. The CTD format simplifies the regulatory review process and underscores a commitment to transparency and scientific rigor.

Key Phases of the NDA Review Timeline
The FDA’s NDA review timeline is structured into distinct phases, each with specific objectives and milestones that collectively determine the fate of a new drug application. To navigate the regulatory landscape with a smooth progression from submission to final decision, here’s what you should expect:
Filing Review (Day 0-60)
The filing review is the first phase of the FDA’s evaluation of a New Drug Application (NDA). During this period, the FDA will review the application to make sure it is complete and all the data and documents are provided. This is critical to verify that the submission meets the minimum requirements for a full evaluation. Within this timeframe, the FDA will also determine if the application is robust enough to be reviewed.
A key milestone in this phase is the Day 74 Letter. This is a communication from the FDA that formally acknowledges the NDA has been filed. The letter informs the sponsor of the application status and the next steps in the review process. The filing of the NDA is a significant indicator that the application meets the minimum requirements for a full evaluation.
Review Clock (Day 60-180)
Once the NDA is accepted, the review clock begins to tick, marking the start of a detailed examination of the submitted data. During this phase, the FDA assigns a multidisciplinary team of experts, including specialists in medicine, pharmacology, and statistics. Each member of the review team plays a critical role in assessing various aspects of the drug’s safety, efficacy, and quality. Their evaluations are essential in forming a holistic understanding of the drug’s benefits and potential risks.
A key component of this phase is the Advisory Committee Meeting. This meeting brings together external experts to provide an independent assessment of the NDA. The committee’s discussions and recommendations can significantly influence the FDA’s final decision. Their input provides an additional layer of scrutiny and ensures that diverse perspectives are considered in the review process.
Action Phase (Day 180-300)
The action phase culminates the NDA review process, leading to a final decision on the drug’s approval status. One of the most important elements of this phase is the PDUFA Date, which stands for the Prescription Drug User Fee Act. The PDUFA establishes a timeline by which the FDA commits to completing its review. This date is a critical benchmark for both the agency and the sponsor, as it provides a target for the final decision.
Upon reaching the PDUFA date, the FDA will issue a decision. The possible outcomes include approval, which allows the drug to enter the market, or a complete response letter (CRL), which indicates that the application cannot be approved in its current form. Alternatively, the FDA may request additional information or studies to resolve outstanding issues. Each of these outcomes has significant implications for the sponsor and the drug’s potential market entry.

Factors Influencing the NDA Review Timeline
Various elements can significantly affect the NDA review timeline, shaping the pace and progression of the evaluation process.
Priority Review vs. Standard Review
One of the most significant factors influencing the NDA review timeline is the designation of the application as either a priority review or a standard review. A priority review designation is granted to drugs that offer significant improvements in the treatment, diagnosis, or prevention of serious conditions compared to available therapies. This designation accelerates the review process, shortening the timeline from the standard ten months to six months, thus expediting the drug’s potential entry into the market.
In contrast, a standard review applies to drugs that do not meet these criteria, following the typical review period.
Breakthrough Therapy Designation
The Breakthrough Therapy Designation is another special status that can significantly expedite the NDA review process. This designation is reserved for drugs that demonstrate substantial improvement over existing therapies in treating serious or life-threatening conditions based on preliminary clinical evidence.
Drugs with this designation benefit from more intensive guidance from the FDA throughout the development process and a more streamlined review. This includes increased communication with FDA officials, the involvement of senior agency staff, and the potential for rolling review, where portions of the application are reviewed as they are submitted rather than waiting for the entire application.
The Path to Approval: Mastering Your Knowledge of NDA Review
The FDA NDA review timeline is an intricate and vital component of the drug approval process. Each phase, from pre-IND meetings to the action phase, is carefully designed to scrutinize every aspect of a drug’s development.
As pharmaceutical companies navigate this complex journey, their understanding of these critical elements will be key to successfully bringing innovative treatments to market, ultimately benefiting patients and advancing medical science.