April 20, 2018 | Kasturi Puranam, PhD | Clinical Research Scientist II | Regulatory Affairs, Drug Development Consulting
I hope that you have read our previous blog posts on IND filing and have learned about the types of INDs, when you need to file an IND, and pre-IND activities. Continue reading this post to understand the process of submitting an initial IND and the subsequent IND-related steps to take as you proceed towards your marketing application.
Let us start with …
IND components
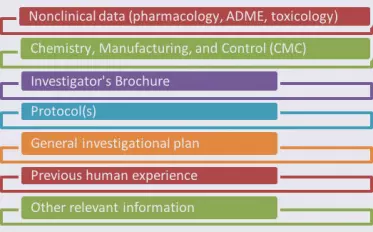
The format and contents of an IND are detailed in 21CFR312. While the IND contents will be specific to your application, the information you submit will fall in categories common to all INDs.
As you prepare the various documents needed for your IND, you will have to keep the CTD hierarchy (“Triangle” or “Pyramid”) in mind.
What is the CTD?
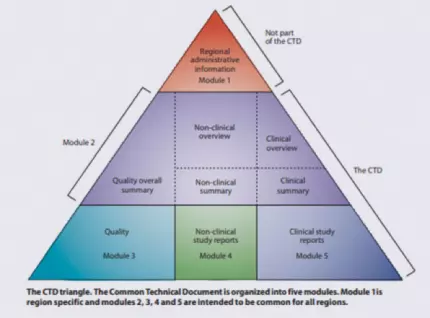 The Common Technical Document (CTD) was developed by the International Council for Harmonisation (ICH) in an attempt to harmonize the marketing application structure and format in the US, EU, Japan, and other countries adhering to ICH guidance. A CTD has five modules: region-specific information (Module 1), summary documents (Module 2), quality-related (CMC) information (Module 3), nonclinical study reports (Module 4), and clinical study reports (Module 5).
The Common Technical Document (CTD) was developed by the International Council for Harmonisation (ICH) in an attempt to harmonize the marketing application structure and format in the US, EU, Japan, and other countries adhering to ICH guidance. A CTD has five modules: region-specific information (Module 1), summary documents (Module 2), quality-related (CMC) information (Module 3), nonclinical study reports (Module 4), and clinical study reports (Module 5).
Historically, submissions to the FDA have been made in paper format. In order to address the issue of storage space and other resources, FDA decided to create electronic submission standards and in 2003 started accepting electronic CTDs (eCTDs). By 2008, eCTDs became the recommended standard for submissions and as of May 2017, eCTDs have been the required format for submissions of marketing applications (to FDA’s Center for Biologics Evaluation and Research [CBER] and Center for Drug Evaluation and Research [CDER]).
Get ready…
 As of May 5, 2018, INDs must be submitted using the eCTD format and FDA will no longer accept any other format (ie, no more paper!). The eCTD webpage is available to help you access a wide variety of resources and support regarding eCTD submissions.
As of May 5, 2018, INDs must be submitted using the eCTD format and FDA will no longer accept any other format (ie, no more paper!). The eCTD webpage is available to help you access a wide variety of resources and support regarding eCTD submissions.

Not ready for this mandatory switch to eCTD? We can help! Contact us.
Publishing and electronic submission considerations
Once your IND is ready, you will file the application using FDA’s Electronic Gateway System (ESG) which is a centralized, agency-wide communication point for receiving secure electronic regulatory submissions. The ESG is a conduit along which submissions travel to reach the proper FDA Center or Office.
Requesting a pre-assigned application number
Every IND, or any other application to FDA, requires an application number. Since IND applications will now be filed electronically (as of May 5), you will need an application number before you file the IND. For this, you will need to request a pre‑assigned application number. Before you make this request, however, you will need to apply for a secure email with FDA. A pre-assigned number will be issued within 3 business days.
If you need help requesting a pre-assigned application number for your IND, contact us.
What happens after your IND is filed?
Once you file an IND, FDA has 30 days to review your application. FDA “inaction” after 30 days have passed means that the study submitted in the IND can proceed. Otherwise, FDA will issue a clinical hold and let you know (more on this below).
 During the 30-day review period, however, FDA can be in communication with you, as the sponsor, to ask for clarifications. Be prepared to provide additional information and/or answer any questions FDA may ask in a timely manner. The types of questions or issues that might arise could center around the suitability of nonclinical studies supporting the IND, manufacturing and other quality issues, or clinical study issues, such as study design, patient population, dosing, safety monitoring plan, etc.
During the 30-day review period, however, FDA can be in communication with you, as the sponsor, to ask for clarifications. Be prepared to provide additional information and/or answer any questions FDA may ask in a timely manner. The types of questions or issues that might arise could center around the suitability of nonclinical studies supporting the IND, manufacturing and other quality issues, or clinical study issues, such as study design, patient population, dosing, safety monitoring plan, etc.
What is a clinical hold?
The FDA review process might conclude that deficiencies exist in the IND, leading to issuance of a clinical hold letter, which is an order by FDA to the sponsor of an IND to delay or suspend the clinical investigation. A “partial” clinical hold will result in a delay or suspension of a particular trial or part of a trial, whereas a “complete” clinical hold will result in a delay or suspension of all clinical trials requested, or currently being conducted, under an IND. Reasons for a clinical hold include, but are not limited to: significant risk to subjects in the clinical study; deficient plan or protocol; insufficient information about the risks in the clinical study; misleading, erroneous, or materially incomplete Investigator’s Brochure; or unqualified clinical investigators.
The clinical hold order may be made by telephone or other means of rapid communication or in writing by (or on behalf of) the Division Director with responsibility for review of the IND. As soon as possible, but no more than 30 days after the clinical hold has been imposed, a written explanation of the  basis of the hold will be issued by FDA and sent to the sponsor. The letter will identify the studies under the IND application to which the hold applies, and will briefly explain the basis for the imposition of the clinical hold. The IND sponsor is expected to address the cited deficiencies in writing and submit a complete response to the issue(s) identified in the clinical hold letter in a separate submission. FDA will review the submission within 30 days. The investigation may resume after FDA has notified the sponsor that the hold has been lifted. The sponsor may disagree with the reasons cited for the clinical hold; in that case, the sponsor should request reconsideration of the decision through the Ombudsman and in accordance with Dispute Resolution procedures.
basis of the hold will be issued by FDA and sent to the sponsor. The letter will identify the studies under the IND application to which the hold applies, and will briefly explain the basis for the imposition of the clinical hold. The IND sponsor is expected to address the cited deficiencies in writing and submit a complete response to the issue(s) identified in the clinical hold letter in a separate submission. FDA will review the submission within 30 days. The investigation may resume after FDA has notified the sponsor that the hold has been lifted. The sponsor may disagree with the reasons cited for the clinical hold; in that case, the sponsor should request reconsideration of the decision through the Ombudsman and in accordance with Dispute Resolution procedures.

IND maintenance
It is important to note that an IND is a “living” document. From FDA’s perspective, the IND phase of drug development spans the time from the initial IND to the submission of a marketing application. Therefore, it is the sponsor’s responsibility to keep the IND updated, or “maintain” the IND. For this, there are three main reporting responsibilities, each of which has a specific structure and deadline for submission. Protocol Amendments and Safety Reports are submitted as needed (to update or change the study plan and to report suspected adverse reactions that are observed in the clinical study, respectively). Annual Reports are submitted every year, even if there are no studies being conducted under the IND.
Here are some details about these three types of reports involved in IND maintenance:
- IND Protocol Amendments: Protocol Amendments are to be submitted to notify FDA of any change to an existing protocol, add a new study protocol, or update investigator information. If the Protocol Amendment is for a new study to be conducted under the IND, there is no 30-day review period; however, an IRB must have approved the new protocol. If adding new investigators or updating investigator information (ie, Form FDA 1572), the investigators’ information may be batched and submitted every 30 days.
- IND Safety Reports: Safety Reports are expedited, written notifications to FDA of an adverse event, considered to be serious, unexpected, and related to study drug administration, or any finding in laboratory animals that would suggest significant risk for human subjects. These reports must be communicated in writing to FDA and all participating investigators within 15 calendar days of the sponsor’s initial receipt of such information. If the event was fatal or life-threatening, FDA is to be notified by telephone or fax within 7 calendar days of receipt of such information. In addition, the sponsor is required to submit 15-day follow-up reports if additional information becomes available after submission of the initial 15-day safety report submission. An IND Safety Report is made on a MedWatch Form (Form FDA 3500A).
- IND Annual Reports: Annual Reports are submitted to notify FDA of the progress of studies conducted under the IND; these are due within 60 days of the anniversary date of the IND becoming active. These reports include, but aren’t limited to: individual study information (eg, patient population, enrollment status, demographics, and discontinuations), brief descriptions of available study reports (clinical and nonclinical investigations), CMC updates, and a general investigational plan for the upcoming year.
To summarize …
When you have all your IND components ready and formatted according to the eCTD structure, you will be ready to submit your IND, electronically, to FDA. Once filed, FDA will assign your application to a reviewing division which will then assign a review team made up of nonclinical, clinical, and CMC experts. An FDA regulatory project manager (RPM) will work with this team and will be your main contact person. Within 30 days after your IND has been filed, unless you have heard otherwise from FDA, your IND becomes effective and the clinical study may proceed.
However, your work with the IND is not over! You will be actively involved in the IND maintenance phase along with the development of your drug all the way up until you file the marketing application. For more information, refer to the FDA guidance on IND applications.
I hope that you have found this final post in our 3-part series on INDs useful. Please contact us if you have any questions or if you need any help with your upcoming IND submission!

Category: Regulatory Affairs, Drug Development Consulting
Keywords: eCTD, clinical hold, IND maintenance
Other Posts You Might Like: