February 25, 2019 | Cheryl Ainslie, PhD, Clinical Research Scientist II | Regulatory Affairs Services

In a previous post, I summarized the process by which the International Council for Harmonisation (ICH) creates harmonized guidelines for use by the pharmaceutical industry. Among the guidelines currently undergoing revision through this process is Efficacy (E) 9, Statistical Principles for Clinical Trials, which was initially released in February 1998. According to ICH, “This biostatistical Guideline describes essential considerations on the design and analysis of clinical trials, especially the ‘confirmatory’ (hypothesis-testing) trials that are the basis for demonstrating effectiveness.”
Statistical Principles for Clinical Trials
Guideline E9 encouraged the use of intention-to-treat analyses, which categorize subjects or patients based on the treatment they were assigned rather than the treatment they actually received or took on their own. This strategy reflects treatment of patients outside of the clinic, where a variety of circumstances may affect how compliant they are with the treatment prescribed.
According to the current Guideline, “subjects allocated to a treatment group should be followed up, assessed and analysed as members of that group irrespective of their compliance to the planned course of treatment…Under many circumstances the full analysis set may also provide estimates of treatment effects likely to mirror those in practice.” However, there was no consensus for how compliance should be measured across clinical trials or treatment regimens as well as for how to adjust for missing data, subject deaths, protocol deviations, non‑adherence to the study drug, or use of prohibited medications.
These issues are collectively referred to as intercurrent events, ie, events that occur after treatment initiation and either preclude observation of an outcome variable or affect its interpretation. The use of estimands can be beneficial in analyzing and interpreting results when intercurrent events occur during the course of a trial. Statistical experts have used the term estimand to refer to a value that is estimated and differs from a parameter in that an estimand can account for intercurrent events. In October 2014, the ICH released a concept paper entitled “Choosing Appropriate Estimands and Defining Sensitivity Analyses in Clinical Trials,” and an addendum to E9 (E9[R1]) began the process of harmonization.
Step 1 (Technical Document) was endorsed by ICH in July 2017, accepted by the European Medicines Association in August 2017, and made available for comment through the US Food and Drug Administration in October 2017. The addendum reached Step 2b (Draft Guideline) in August 2017, and the deadline for comments was 30 April 2018 in the US. Currently, the addendum is in Step 3 (Regulatory Discussion), and the Expert Working Group continues to hold meetings to revise and clarify the document as well as remove redundancies.
What is an estimand?
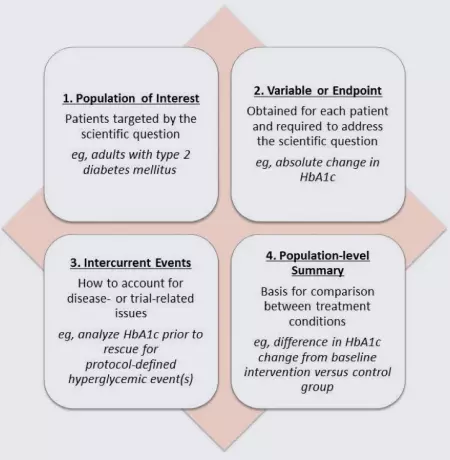
Although not yet common in most regulatory writing, with the likely endorsement of E9(R1), medical writers may begin to use the term estimand more in the development of clinical study protocols, statistical analysis plans, clinical study reports, and integrated summaries. According to the draft addendum, an estimand comprises the 4 factors shown in the figure below. Each of these factors is essential to measuring the primary outcome of a study, and all 4 are required to define the outcome while accounting for intercurrent events.
In this example, the primary outcome is the change in glycosylated hemoglobin (HbA1C) in patients with type 2 diabetes mellitus. In this population, protocol-defined use of a rescue medication for hyperglycemic events is common. Rescue medication is an example of an intercurrent event that can affect the interpretation of a study because it often directly impacts the measurement of the primary outcome variable.
An example estimand based on the 4 components described above is shown below.

A few more examples of estimands:
- Difference in the proportion of patients with type 2 diabetes mellitus with HbA1c <7% at Week 48 in the treatment group versus a control group regardless of rescue for protocol-defined hyperglycemic events
- Mean change from baseline to Week 48 in HbA1C in the treatment group versus a control group regardless of rescue for protocol-defined hyperglycemic events in patients with type 2 diabetes mellitus
- Difference in the time for patients with type 2 diabetes mellitus to achieve HbA1c <7% without receiving rescue for protocol‑defined hyperglycemic events in the treatment group versus a control group
Estimands in protocol writing
According to the E9(R1) guideline, clinical trial protocols should establish the primary estimand that aligns with the primary objective of the trial: “Estimands should be defined and explicitly specified in the clinical trial protocol.” The guidance includes a few other pointers for protocol development:
- Generating estimands is multidisciplinary and may involve clinicians, statisticians, and regulatory experts
- Trial design should be based on the choice of an estimand or estimands that reflect the primary trial objective(s)
- Intercurrent events should be anticipated and explicitly specified
- The protocol should identify the estimator associated with the primary estimand. An estimator is the analytic approach to compute an estimate (ie, population-level summary) from the observed clinical trial data, eg, mean change from baseline in HbA1C computed via analysis of covariance

During protocol development is also an excellent time to plan for adequate sensitivity analyses to account for intercurrent events. These analyses should be included to measure the robustness of the primary estimand, ie, if the statistical models are still valid if the data violate the underlying assumptions (eg, are not normally distributed). Sometimes, the use of certain estimands or sensitivity analyses may require the use of nonstandard trial designs, such as run-in or titration designs.
A schematic for the planned estimand with main estimator and estimate is shown below, followed by examples including the accompanying sensitivity analyses.
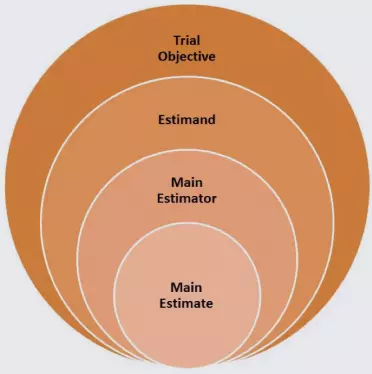
- Trial Objective
- To determine the effectiveness of the treatment to reduce HbA1C in patients with type 2 diabetes mellitus
- Estimand
- Mean change from baseline to Week 48 in HbA1C in the treatment group versus a control group prior to rescue for protocol-defined hyperglycemic events in patients with type 2 diabetes mellitus
- Main Estimator
- Mean change from baseline in HbA1C computed via analysis of covariance
- Sensitivity Estimator 1
- Missing data imputed via last observation carried forward
- Sensitivity Estimator 2
- Analysis including observed values only
- Main Estimate
- Patients with type 2 diabetes mellitus who complete 48 weeks of treatment have a mean decrease in HbA1C of X%.
- Sensitivity Estimate 1
- Patients with type 2 diabetes mellitus who complete 48 weeks of treatment have a mean decrease in HbA1C of X%, where missing data were imputed using the last observation carried forward method.
- Sensitivity Estimate 2
- In all patients with type 2 diabetes mellitus with HbA1C measurements collected according to the protocol, there was a mean decrease in HbA1C of X% at Week 48.
- Sensitivity Estimate 1
- Patients with type 2 diabetes mellitus who complete 48 weeks of treatment have a mean decrease in HbA1C of X%.
- Sensitivity Estimator 1
- Mean change from baseline in HbA1C computed via analysis of covariance
Secondary objectives of a study are not required to have estimands by the E9(R1) guideline, unless the objectives are intended to support regulatory decisions. In this case, each objective should be described fully with a corresponding main estimator and suitable sensitivity analysis. Other secondary and exploratory objectives do not need to have estimands, but if they address scientific questions of interest that are substantially different from the question addressed by the primary objective, it is recommended that these be fully documented in the protocol, potentially using different estimands.
An example would be changes in patient-reported outcome measurements of diabetic neuropathy symptoms. Because this endpoint is different from changes in HbA1C, a detailed description of the estimand(s) and specified sensitivity analyses may be beneficial to interpreting the data collected.
Estimands in statistical analysis planning

For transitioning the protocol-planned clinical study design to a statistical analysis plan, the E9(R1) guideline also includes guidance for ensuring estimands are properly measured and analyzed. Sample size calculations should be adjusted to account for the intercurrent events that will be included in the analysis of the primary estimand. In addition, when there is a plan to pool data across multiple clinical trials in the same program, each clinical trial should incorporate appropriate estimands to allow for pooling for analysis.
The E9(R1) guideline states that all data should be collected that are needed to analyze the primary estimands, even if they are measured after intercurrent events. If there are data that were not collected to assess an estimand, collecting and reporting the reasons why data were not collected can help distinguish intercurrent events from missing data, which would provide more information about the primary objective during the statistical analysis of the estimand.
By collecting and reporting the reasons for missing data, the statistical analysis plan can include the appropriate imputation methods and sensitivity analyses. For example, a subject may have been lost to follow up due a random reason (ie, not related to a variable being measured), such as moving out of state, or due to a nonrandom reason (ie, related to a variable being measured), such as withdrawing consent because of perceived lack of efficacy of the treatment being studied.
If data are missing at random (as in the first example), a sensitivity analysis may be designed to compare the results from an analysis including all observed values with an analysis that imputes missing data using the last observation carried forward or a likelihood‑based mixed‑effects analysis. If data are missing not at random (as in the second example), a sensitivity analysis may compare the results from all observed values with the results from an analysis that imputes missing data using the nonresponder imputation.
In a large clinical trial, it is likely that several imputation methods and a variety of sensitivity analyses will be needed to assess the robustness of the final data given the intercurrent events identified in the protocol. This strategy ultimately provides more information for what may happen when the treatment is used outside of a clinical trial setting compared with treating all missing data as equivalent.
Assumptions for each analysis should be stated explicitly and be justifiable and plausible. For example, the main analysis could assume that for any subject who died before the last assessment (an intercurrent event), the value for the assessment from the most recent previous assessment could be imputed, ie, last observation carried forward. A sensitivity analysis would be conducted to demonstrate the robustness of the estimate from the analysis with this assumption.
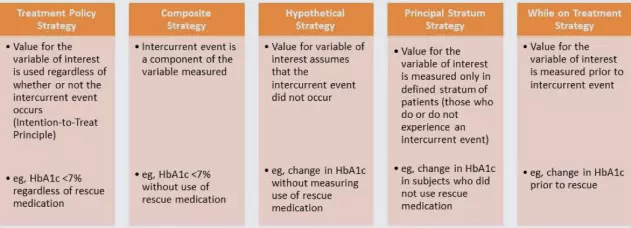
The E9(R1) guideline offers some potential strategies to address intercurrent events during analysis, which are shown below. Each type of intercurrent event would require its own strategy.
Estimands in clinical study reports
The E9(R1) guideline states that clinical study reports should present the results from the main, sensitivity, and any supplementary analyses systematically, and it should be stated whether each analysis was prespecified, introduced while the trial was still blinded, or post hoc. The reports should discuss any intercurrent events that were not foreseen or considered during trial design—how these were accounted for (or not) during analyses and what impact their accounting may have on the estimates and their interpretation.
The formulation of specific and appropriate estimands during the earliest stages of trial planning can help limit the ambiguity of clinical trial results. This may streamline the clinical trial process to bring new, effective treatments to market more quickly.
Getting our statistics in tune

With its anticipated adoption as an ICH Harmonized Guideline, E9(R1) presents an exciting opportunity for improved data collection and reporting that more accurately reflects the behaviors of patients in their “real world,” daily life. By collecting and reporting data that realistically reflect patients’ behavior in taking a medication or using a medical device, pharmaceutical companies will be able to offer more effective therapies in less time than was needed for trials in the past.
Will you need to revise or compose documents to follow the E9(R1) guideline? IMPACT has experienced regulatory affairs professionals to assist you with incorporating the E9(R1) guideline when preparing high-quality regulatory documents. If you have any questions about the E9(R1) guideline or would like to work with us, please do not hesitate to contact us.

Category: Regulatory Affairs Services
Keywords: ICH; Statistics; Estimands; Harmonization; Intercurrent events; Clinical Study Protocol; Clinical Study Report; Clinical trials; Statistical Analysis Plan
Other Posts You Might Like: